10 September 2020
Originally published via the University of Melbourne by Stephanie Portelli, Elston D’Souza and Associate Professor David Ascher
COVID-3D, a new open-access tool harnesses SARS-CoV-2 genomic and structural information to help ensure therapeutic efforts remain effective.
Eight months ago, on the 31st of December 2019, the first case of COVID-19 was reported in Wuhan in China.
Since then, the coronavirus SARS-CoV-2 has caused the biggest global pandemic since the 1918/1919 Spanish Flu.
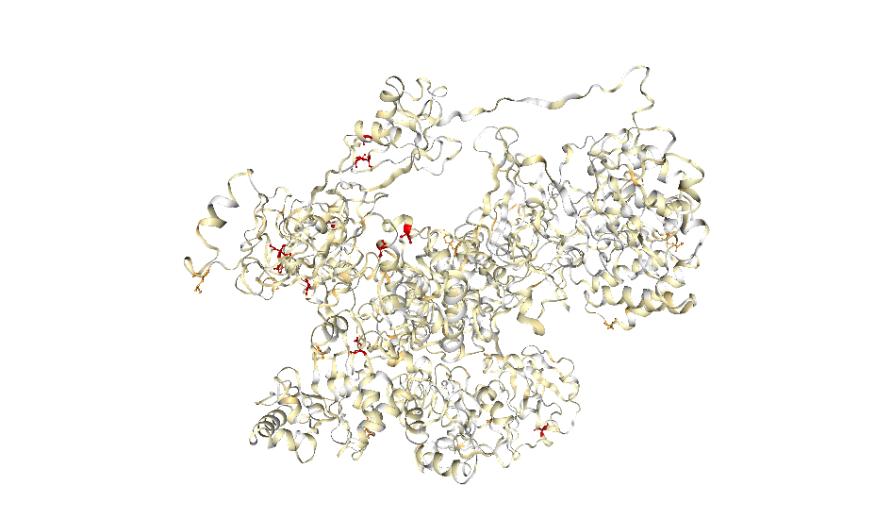
As of early September this year, coronavirus infections have exceeded 22 million and caused more than 776,000 deaths. And despite its seclusion, Australia has also been impacted with more than 26,000 confirmed cases and over 700 deaths.
Currently, there are around 34 vaccine candidates and more than 500 drugs undergoing clinical trials. But a considerable challenge is not just ensuring that our vaccines and drugs work, but that they remain effective as the virus mutates.
Although the virus SARS-CoV-2 is a relatively new pathogen, its ability to readily accumulate mutations across its genes was evident from the start of this pandemic.
In the context of therapeutic drug design and discovery, these mutations, and the patterns with which they accumulate within the virus’ protein structures, might affect the ability of vaccines and drugs to bind the virus, or to create a specific immune response against it.
As a result, as scientists, we not only need to try to control the virus, but we need to outsmart it by predicting how it will change over time.
SARS-COV-2 Mutational Consequences
Mutations, or changes in an organism’s genetic material, are natural effects of evolution after replication.
Although these changes can be viewed as ‘errors’ in the replication process, mutations occurring in the ‘right’ place can give the virus new powers, making them ‘necessary evils’ for viral survival.
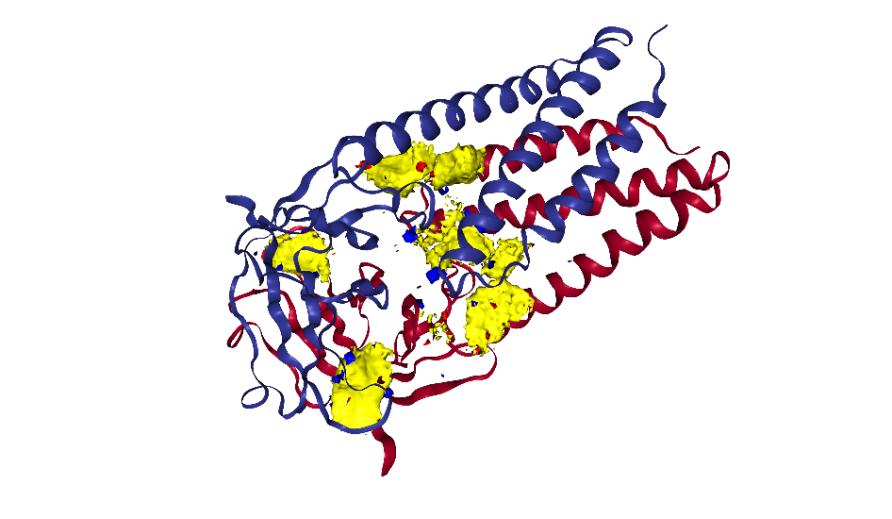
The Spike protein D614G mutation, for instance, which was first seen in Europe and now across the globe, has made SARS-CoV-2 more transmittable.
Luckily, here in Australia, we have seen strains of the virus that are predicted to be less severe than in other countries around the world.
Biologically, viral populations that contain these advantageous mutations are more likely to prosper and replicate and are therefore more likely to be found in new infections. This leads to the mutations becoming ‘fixed’ within the population.
Fortunately, SARS-CoV-2 is mutating slower than other viruses, like influenza, with about two new changes in its genome every month.
So, what happens when a SARS-CoV-2 gene is mutated?
Each gene is aided by the cell’s machinery within the host organism to make a specific protein. Unlike DNA or RNA, which are like wound-up pieces of ribbon, proteins have three dimensional structures; their surfaces appearing like mountains, valleys, ridges, pockets and grooves.
The resultant mutant protein usually has a slightly different shape than the normal protein, particularly around the area where the mutation lies.
For example, if a protein is mutated at the site where another protein or drug would usually bind, this interaction might be compromised.
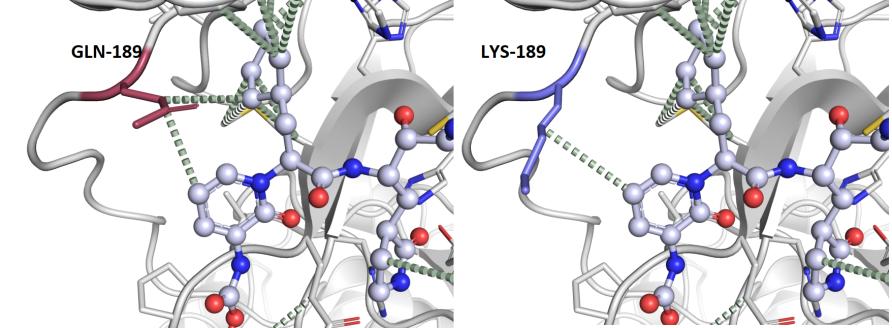
Because therapies like vaccines and drugs target proteins specifically, mutations that compromise drug interactions present a major challenge in therapeutic drug development, especially as new mutations may render some of these therapies ineffective over time.
Modelling Mutational Hotspots
What if researchers could predict these ‘mutational hotspots’, as well as their effects on the shape of the protein and function? Then scientists could direct their efforts towards biologically important but less mutated regions – more of a guided-missile strategy.
Failure to do so, would keep the virus in circulation, lead to more outbreaks and keep scientists chasing it, one step behind its newest mutation.
To aid these therapeutic efforts, our team at the Bio21 Molecular Science and Biotechnology Institute and the Baker Heart and Diabetes Institute, have developed COVID-3D, a software tool and library.
This contains information about all the protein structures that coincide with the whole SARS-CoV-2 genome including every known genetic mutation and its resultant mutant protein structure. The tool can also be used to help researchers recognise when mutations affect drug binding or to identify alternative targets for drug and vaccine development.
To develop this tool, our team analysed the genome sequencing data of over 120,000 samples of SARS-CoV-2 from infected people around the globe (including those that uniquely affect Australia) to identify mutations present within each protein. Our research was published in Nature Genetics.
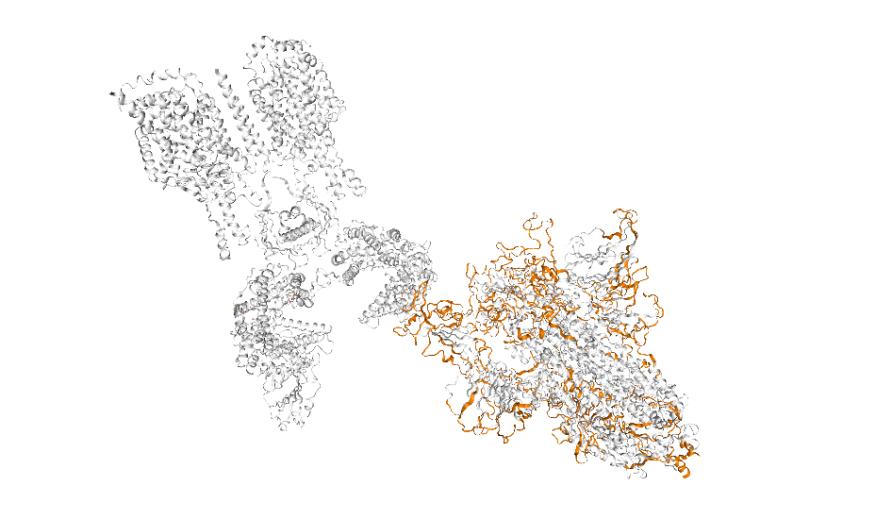
We then tested and analysed the effects of these clinically observed mutations on their protein structure, and also looked at the effects of every possible mutation within the genome.
Staying A Step Ahead
SARS-CoV-2 genome sequencing has revealed its genetic relatedness to two distinct coronaviruses: the disease-causing SARS-CoV and the RaTG13 virus, which infects bats.
As both these viruses have been circulating for years, this relatedness can be exploited to anticipate how the novel virus will mutate over time. Analyses of these mutations are also provided in COVID-3D to help researchers account for these possible future mutations.
All mutations can be easily visualised on the protein structure, which can be coloured according to specific protein properties – like how conserved the structure is in evolution.
This helps researchers to locate structural ‘weak-points’, which can serve as targets for more effective, longer-lasting therapies, despite any new mutations.
As the global scientific and medical community continuously gains better understanding of the biology behind SARS-CoV-2 infection and disease, this will be a powerful resource to guide the development of more tailored therapies.
COVID-3D is continuously being updated with new protein structures, mutations, and analyses to help keep increasing our understanding of the SARS-CoV-2 mechanisms of disease.


